Cadiot-Chodkiewicz Coupling

The copper(I)-catalyzed coupling of a terminal alkyne and an alkynyl halide offers access to unsymmetrical bisacetylenes.
Mechanism of the Cadiot-Chodkiewicz Coupling

====================================
Cannizzaro Reaction

This redox disproportionation of non-enolizable aldehydes to carboxylic acids and alcohols is conducted in concentrated base.
α-Keto aldehydes give the product of an intramolecular disproportionation in excellent yields.

Mechanism of the Cannizzaro Reaction

An interesting variant, the Crossed Cannizzaro Reaction, uses formaldehyde as reducing agent:

At the present time, various oxidizing and reducing agents can be used to carry out such conversions (with higher yields), so that today the Cannizzaro Reaction has limited synthetic utility except for the abovementioned conversion of α-keto aldehydes.
The Cannizzaro Reaction should be kept in mind as a source of potential side products when aldehydes are treated under basic conditions.
====================================
Corey-Bakshi-Shibata Reduction
Itsuno-Corey Reduction

The enantioselective reduction of ketones using borane and a chiral oxazaborolidine as catalyst (CBS catalyst). Usually, MeCBS is used (R'' = Me, but selectivity may be increased by varying this substituent).
Mechanism of the Corey-Bakshi-Shibata Reduction
The mechanism depicted portrays the rationale for the enantioselectivity and high reaction rates, which are influenced only by the CBS catalyst. This catalyst is a combination of both a Lewis acid and a chiral auxiliary!
===================================
Chan-Lam Coupling

This reaction allows aryl carbon-heteroatom bond formation via an oxidative coupling of arylboronic acids, stannanes or siloxanes with N-H or O-H containing compounds in air. Substrates include phenols, amines, anilines, amides, imides, ureas, carbamates, and sulfonamides. The reaction is induced by a stoichiometric amount of copper(II) or a catalytic amount of copper catalyst which is reoxidized by atmospheric oxygen.
The Chan-Lam Coupling may be conducted at room temperature in air, which gives it a certain advantage over the Buchwald-Hartwig Cross Coupling.
Mechanism of the Chan-Lam Coupling
The reaction with a stoichiometric amount of copper(II) is also facilitated by oxygen, because reductive elimination from a copper(III) species is faster.
====================================
Claisen Condensation
 The Claisen Condensation between esters containing α-hydrogens, promoted by
a base such as sodium ethoxide, affords β-ketoesters. The driving force
is the formation of the stabilized anion of the β-keto ester. If two
different esters are used, an essentially statistical mixture of all
four products is generally obtained, and the preparation does not have
high synthetic utility.
The Claisen Condensation between esters containing α-hydrogens, promoted by
a base such as sodium ethoxide, affords β-ketoesters. The driving force
is the formation of the stabilized anion of the β-keto ester. If two
different esters are used, an essentially statistical mixture of all
four products is generally obtained, and the preparation does not have
high synthetic utility.However, if one of the ester partners has enolizable α-hydrogens and the other does not (e.g., aromatic esters or carbonates), the mixed reaction (or crossed Claisen) can be synthetically useful. If ketones or nitriles are used as the donor in this condensation reaction, a β-diketone or a β-ketonitrile is obtained, respectively.
The use of stronger bases, e.g. sodium amide or sodium hydride instead of sodium ethoxide, often increases the yield.
The intramolecular version is known as Dieckmann Condensation.
Mechanism of the Claisen Condensation



===================================
Claisen Rearrangement

The aliphatic Claisen Rearrangement is a [3,3]-sigmatropic rearrangement in which an allyl vinyl ether is converted thermally to an unsaturated carbonyl compound.
The aromatic Claisen Rearrangement is accompanied by a rearomatization:

The etherification of alcohols or phenols and their subsequent Claisen Rearrangement under thermal conditions makes possible an extension of the carbon chain of the molecule.
Mechanism of the Claisen Rearrangement
The Claisen Rearrangement may be viewed as the oxa-variant of the Cope Rearrangement: Mechanism
of the Cope Rearrangement
Mechanism
of the Cope Rearrangement Mechanism
of the Claisen Rearrangement
Mechanism
of the Claisen RearrangementThe reaction proceeds preferably via a chair transition state. Chiral, enantiomerically enriched starting materials give products of high optical purity.

A boat transition state is also possible, and can lead to side products:

The aromatic Claisen Rearrangement is followed by a rearomatization:

When the ortho-position is substituted, rearomatization cannot take place. The allyl group must first undergo a Cope Rearrangement to the para-position before tautomerization is possible.

All Claisen Rearrangement reactions described to date require temperatures of > 100 °C if uncatalyzed. The observation that electron withdrawing groups at C-1 of the vinyl moiety exert a positive influence on the reaction rate and the yield has led to the development of the following variations:
 Ireland-Claisen Rearrangement
Ireland-Claisen Rearrangement Eschenmoser-Claisen Rearrangement
Eschenmoser-Claisen Rearrangement Johnson-Claisen
Rearrangement
Johnson-Claisen
Rearrangement=====================================
Clemmensen Reduction

The Clemmensen Reduction allows the deoxygenation of aldehydes or ketones, to produce the corresponding hydrocarbon.
The substrate must be stable to strong acid. The Clemmensen Reduction is complementary to the Wolff-Kishner Reduction, which is run under strongly basic conditions. Acid-labile molecules should be reduced by the Wolff-Kishner protocol.
Mechanism of the Clemmensen Reduction
The reduction takes place at the surface of the zinc catalyst. In this reaction, alcohols are not postulated as intermediates, because subjection of the corresponding alcohols to these same reaction conditions does not lead to alkanes. The following proposal employs the intermediacy of zinc carbenoids to rationalize the mechanism of the Clemmensen Reduction:
=====================================
Cope Elimination

The Cope Reaction of N-oxides, which can easily be prepared in situ from tertiary amines with an oxidant such as peracid, leads to alkenes via a thermally induced syn-elimination in aprotic solvents.
Mechanism of the Cope Elimination
The Cope Elimination is a syn periplanar elimination in which six electrons move in a five-membered ring according to a concerted, thermally-induced mechanism to yield an alkene and a hydroxylamine:
The sterically demanding amine oxide function reacts preferentially with the more easily accessible hydrogens, and often gives good selectivity favoring the less-substituted alkene. Thus, for simple alkenes, the reaction follows the Hofmann Rule.
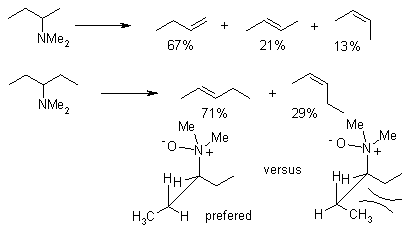
The following structures exemplify the stereochemical requirement for reaching a five-membered cyclic transition state and the influence of sterically demanding groups for some cyclic compounds:

For an early review on the Cope Elimination and similar reactions showing more examples, please refer to DePuy and King (Chem. Rev. 1960, 60, 432. DOI).
One synthetically useful exception to the general preference for more accessible hydrogens is the reaction of substrates that bear a β-phenyl group, or more generally speaking, an electron-withdrawing group in the β-position:

Compared with alkyl-substituted derivatives, a phenyl group provides a 100-fold rate increase. Phenyl and other electron-withdrawing groups lower the electron density of the carbon-hydrogen bond, making the hydrogen more acidic, and stabilizing the transition state. Computed minimal energy paths suggest that the Cope Elimination is slightly dissymmetric and nonsynchronous, with the H-transfer occurring slightly in advance of the other bond reorganizations (I. Komaromi, J. M. Tronchet, J. Phys. Chem. A 1997, 101, 3554).
Another interesting feature is the solvent dependence of the reaction rate. The Cope Elimination is extraordinarily sensitive to solvent effects, and a million-fold rate increase can be obtained going from protic to aprotic solvents. Within aprotic solvents, decreasing polarity significantly increases the reaction rate. The relative rate retardation for Cope Eliminations in protic solvents arises from hydrogen bonding between the amine oxide and the solvent. In addition, a fully solvated amine oxide in a protic solvent such as MeOH can even be relatively more favorable energetically than the less polar hydroxylamine, which can displace the equilibrium to favor the retro-Cope Elimination (O. Acevedo, W. L. Jorgensen, J. Am. Chem. Soc. 2006, 101, 6141):

====================================
Cope Rearrangement
(Anionic)
Oxy-Cope Rearrangement
The Cope Rearrangement is the thermal isomerization of a 1,5-diene
leading to a regioisomeric 1,5-diene. The main product is the
thermodynamically more stable regioisomer. The Oxy-Cope has a hydroxyl
substituent on an sp3-hybridized carbon of the starting
isomer.
The driving force for the neutral or anionic Oxy-Cope Rearrangement is that the product is an enol or enolate (resp.), which can tautomerize to the corresponding carbonyl compound. This product will not equilibrate back to the other regioisomer.

The Oxy-Cope Rearrangement proceeds at a much faster rate when the starting alcohol is deprotonated, e.g. with KH. The reaction is then up to 1017 times faster, and may be conducted at room temperature. Aqueous work up then gives the carbonyl compound.

Mechanism of the Cope Rearrangement


Two transition states are possible, and the outcome of the reaction can be predicted on the basis of the most favorable overlap of the orbitals of the double bond, as influenced by stereoelectronic factors:


===================================
Conia-Ene Reaction

The Conia-Ene reaction is an intramolecular, thermal or Lewis acid-catalysed reaction of unsaturated carbonyl compounds to yield cyclised products.
Mechanism of the Conia-Ene Reaction
In the Conia-Ene reaction, enolisation is followed by a concerted 1,5-hydrogen shift:
Thus, the Conia-Ene Reaction is an intramolecular variant of the generally intermolecular Ene Reaction.
Reactions that generate cyclopentane and cyclohexane derivatives in good yields normally proceed at 350°C, but medium-sized rings need higher temperatures and the yield is considerably lower.

Each step of the Conia-Ene reaction is reversible, as shown by some retro-Conia Ene reactions of cyclobutanes where the main product is the open-chain isomer.

The stereochemistry of the cyclisation is affected by the spatial arrangement of the two reactive centres, and this mainly depends on the configuration at the enol centre and the various steric effects arising from the chain conformation in the ground state.

In cases where the cyclic product is still enolisable, subsequent enolisation results in epimerisation that can modify the product ratio and therefore no simple conclusion regarding the stereoselectivity can be reached.
The scope of substrates includes precursors of ketones such as acetals and enol ethers, which also regenerate the ketone at high temperature. However, unsaturated β-ketoesters and β-diketones are also suitable starting materials. Their stronger enolic character enables reactions to proceed at lower temperatures.

For acetylenic substrates, double bond migration often occurs to favour a higher degree of substitution. No such migration is observed when a terminal methyl group stabilises the exo-double bond, or if the reaction is conducted at lower temperatures - for example with a β-diketone as substrate.

For a review of thermal Conia-Ene reactions and a detailed mechanistic discussion, please refer to a review by Conia and Le Perchec (Synthesis 1975, 1. DOI)
Nevertheless, the need for high temperatures severely limits the synthetic utility of the Conia-Ene reaction. A catalytic version that proceeds at ambient temperature and under neutral conditions would dramatically increase the reaction’s usefulness. In 2004, Toste reported a phosphinegold(I)-catalysed version for the intramolecular addition of a β-ketoester to an unactivated alkyne. For this reaction, two mechanistic hypotheses have been developed.

J. J. Kennedy-Smith, S. T. Staben, F. D. Toste, J. Am. Chem. Soc., 2004, 126, 4526-4527.
For this system, the deuterium-labelling experiments depicted below supported a mechanism involving enol addition to an Au-alkyne complex (mechanism A above).

Some recent developments for catalysed reactions, including enantioselective reactions, can be found in the recent literature section.
====================================
Corey-Chaykovsky Reaction
The reaction of sulfur ylides with carbonyl compounds such as ketones or the related imines leads to the corresponding epoxides or aziridines.
Corey-Chaykovsky Epoxidation

Corey-Chaykovsky Aziridination
The reaction of sulfur ylides with enones gives cyclopropanes.

Corey-Chaykovsky Cyclopropanation
Mechanism of the Corey-Chaykovsky Reaction
The ylides are generated in situ by the deprotonation of sulfonium halides with strong bases.

Dimethyloxosulfonium methylide - known as the Corey-Chaykovsky Reagent - is a valuable alternative to dimethylsulfonium methylide and can be generated from trimethylsulfoxonium iodide.

Higher substituted ylides can be generated selectively if one substituent is preferably deprotonated over the others, for example when the negative charge is stabilized or the environment is sterically less demanding:

Such ylides are able to transfer more than just a methylene group, and enantioselective induction can be observed if the ylide is chiral:

The ylide initially acts as a nucleophile toward the carbonyl compound. The resulting oxygen anion then reacts as an intramolecular nucleophile toward the now electrophilic ylide carbon, which bears a sulfonium cation as a good leaving group:

The reaction of the Corey-Chaykovsky Reagent with enones is a 1,4-addition that is followed by ring closure to give a cyclopropane:

As sulfides are readily alkylated, it is even possible to use them catalytically. Such methods can give very interesting results when expensive chiral sulfides are used for the generation of chiral epoxides.

For a review of enantioselective methods see: V. K. Aggarwal, J. Richardson, Chem. Commun. 2003, 2644. DOI
With phosphorus ylides as used for the Wittig Reaction, the phosphorus atom forms a strong double bond with oxygen. This leads the mechanism in a different direction, to effect olefination instead of epoxidation through intermediate oxaphosphetanes
====================================
Corey-Fuchs Reaction

This two step methodology allows the preparation of terminal alkynes by one-carbon homologation of an aldehyde. The first step is comparable to a Wittig Reaction, and leads to a dibromoalkene. Treatment with a lithium base (BuLi, LDA) generates a bromoalkyne intermediate via dehydrohalogenation, which undergoes metal-halogen exchange under the reaction conditions and yields the terminal alkyne upon work-up.
A modification of the Corey-Fuchs Reaction involves the reaction of the intermediate alkynyllithium with an electrophile prior to aqueous work-up, giving a chain extension product:

Mechanism of the Corey-Fuchs Reaction
In the formation of the ylide from CBr4, two equivalents of triphenylphosphine are used. One equivalent forms the ylide while the other acts as reducing agent and bromine scavenger.
The addition of the ylide to the aldehyde:

Reaction of the dibromoalkene with BuLi:

====================================
Corey-Kim Oxidation
The Corey-Kim Oxidation allows the synthesis of aldehydes and ketones from primary alcohols and secondary alcohols, respectively.
Mechanism of the Corey-Kim Oxidation
Dimethylchlorosulphonium ion is generated in situ from NCS and DMS:
The following steps are comparable to the Swern Oxidation:



=====================================
Corey-Seebach Reaction
Seebach Umpolung

The Corey-Seebach Reaction uses lithiated 1,3-dithianes as nucleophilic acylating agents.
Mechanism of the Corey-Seebach Reaction
The Corey-Seebach Reaction allows a reversal of the normal reactivity of acyl carbon atoms, which combine only with nucleophiles. The German term "Umpolung" is widely used for this inversion of reactivity.
The lithiated 1,3-dithiane can be viewed as an masked acyl anion that is able to react with various electrophiles.
The acidity difference of hydrogen atoms adjacent to divalent sulfur compared to oxygen stems from the greater polarizability of sulfur and the longer C-S-bond length; d-orbitals are not involved. In most cases treatment of dithianes with n-BuLi at temperatures of -30 °C is sufficient for the preparation of the lithio-derivatives. With pKA values of approximately 30, lithiated dithianes can react with aldehydes or ketones, epoxides and acid derivatives, but also with alkyl halides without competing elimination reactions.

Umpolung offers access to a wide range of products, especially 1,2-diketones and α-hydroxy ketones, products that cannot be obtained using the normal reactivity (for example through aldol addition).
Among the other thioacetals that could be used for Umpolung, metallated dithiolanes undergo fragmentation and disproportionate to ethene and dithiocarboxylates:

1,3-Dithianes are readily prepared from aldehydes (for an overview, see 1,3-dithianes as protecting group) and offer high stability towards acids and bases. Therefore, use of the S,S-acetal unit is especially useful in multistep synthesis. A crucial step is the hydrolysis of S,S-acetals, the difficulty of which is due to the excellent nucleophilicity of sulfur.

Only irreversible removal of the dithiol or of the solvolysis products can push the equilibrium to the right. Methods of choice are transacetalization to a highly reactive carbonyl derivative, alkylation to sulfide, oxidation of the thiol (for recent methods see deprotection of 1,3-dithianes) and formation of metal thiolates, for which mercury(II) salts are frequently used.
For a review on "Umpolung of the Reactivity of Carbonyl Compounds Through Sulfur-Containing Reagents" please check out a publication by Seebach (Synthesis 1977, 357. DOI).
====================================
Corey-Winter Olefin Synthesis

Conversion of 1,2-diols to alkenes. The cyclic thiocarbonate is available from reaction of the diol with thiophosgene or thiocarbonyldiimidazole, and reacts with added trimethylphosphite via a syn-elimination to the alkene.

Mechanism of the Corey-Winter Olefin Synthesis
It is assumed that the reaction proceeds with attack of phosphite on sulphur leading to a carbene, which may react with a second equivalent of phosphite during the cycloreversion that splits out carbon dioxide and yields the product alkene.
=====================================
Pechmann Condensation
Coumarin Synthesis

The Pechmann Condensation allows the synthesis of coumarins by reaction of phenols with β-keto esters.
Mechanism of the Pechmann Condensation
The reaction is conducted with a strong Brønstedt acid such as methanesulfonic acid or a Lewis acid such as AlCl3. The acid catalyses transesterification as well as keto-enol tautomerisation:
A Michael Addition leads to the formation of the coumarin skeleton. This addition is followed by rearomatisation:

Subsequent acid-induced elimination of water gives the product:

====================================
Ozonolysis
Criegee Mechanism



Ozonolysis allows the cleavage of alkene double bonds by reaction with ozone. Depending on the work up, different products may be isolated: reductive work-up gives either alcohols or carbonyl compounds, while oxidative work-up leads to carboxylic acids or ketones.
Mechanism of Ozonolysis
The mechanism was suggested by Criegee (Angew. Chem. Int. Ed., 1975, 87, 745. DOI) and has been recently revisited using 17O-NMR Spectroscopy by the Berger Group (Eur. J. Org. Chem., 1998, 1625. DOI).First step is a 1,3-dipolar cycloaddition of ozone to the alkene leading to the primary ozonide (molozonide, 1,2,3-trioxolane, or Criegee intermediate) which decomposes to give a carbonyl oxide and a carbonyl compound:

The carbonyl oxides are similar to ozone in being 1,3-dipolar compounds, and undergo 1,3-dipolar cycloaddition to the carbonyl compounds with the reverse regiochemistry, leading to a mixture of three possible secondary ozonides (1,2,4-trioxolanes):



These secondary ozonides are more stable than primary ozonides. Even if the peroxy bridge is shielded by steric demanding groups leading to isolable products, they should not be isolated from an unmodified ozonolysis, because still more explosive side products (tetroxanes) may have been formed:

As endoperoxides are investigated as antimalarial compounds, more selective methods have been developed for their preparation (for example the Griesbaum Coozonolysis). Some reactions can be found here: V. D.B. Bonifacio, Org. Chem. Highlights 2004, October 25.
The Criegee mechanism is valid for reactions in hydrocarbons, CH2Cl2, or other non-interactive solvents. Alcohols react with the carbonyl oxide to give hydroperoxy hemiacetals:

The synthetic value lies in the way the complex mixtures of intermediates can be worked up to give a defined composition of products and a clean conversion of all peroxide species. The three main possibilities are given above, along with examples for the reagents used.
====================================
Cross Metathesis
The transalkylidenation of two terminal alkenes under release of ethene, catalyzed by ruthenium carbenoids (Grubbs Catalyst). Statistically, the reaction can lead to three possible pairs of geometric isomers, i.e. E/Z pairs for two homocouplings and the cross-coupling (R-CH=CH-R, R'-CH=CH-R', and R-CH=CH-R') - a total of 6 products.
The selectivity of this reaction is currently undergoing further study, but various examples exist in which two alkenes with different reactivity give the cross-coupled product with excellent yields and excellent selectivity.\
=====================================
Curtius Rearrangement

The Curtius Rearrangement is the thermal decomposition of carboxylic azides to produce an isocyanate. These intermediates may be isolated, or their corresponding reaction or hydrolysis products may be obtained.
The reaction sequence - including subsequent reaction with water which leads to amines - is named the Curtius Reaction. This reaction is similar to the Schmidt Reaction with acids, differing in that the acyl azide in the present case is prepared from the acyl halide and an azide salt.

Mechanism of the Curtius Rearrangement
Preparation of azides:
Decomposition:

Reaction with water to the unstable carbamic acid derivative which will undergo spontaneous decarboxylation:


Isocyanates are versatile starting materials:

Isocyanates are also of high interest as monomers for polymerization work and in the derivatisation of biomacromolecules.
No comments:
Post a Comment